2024
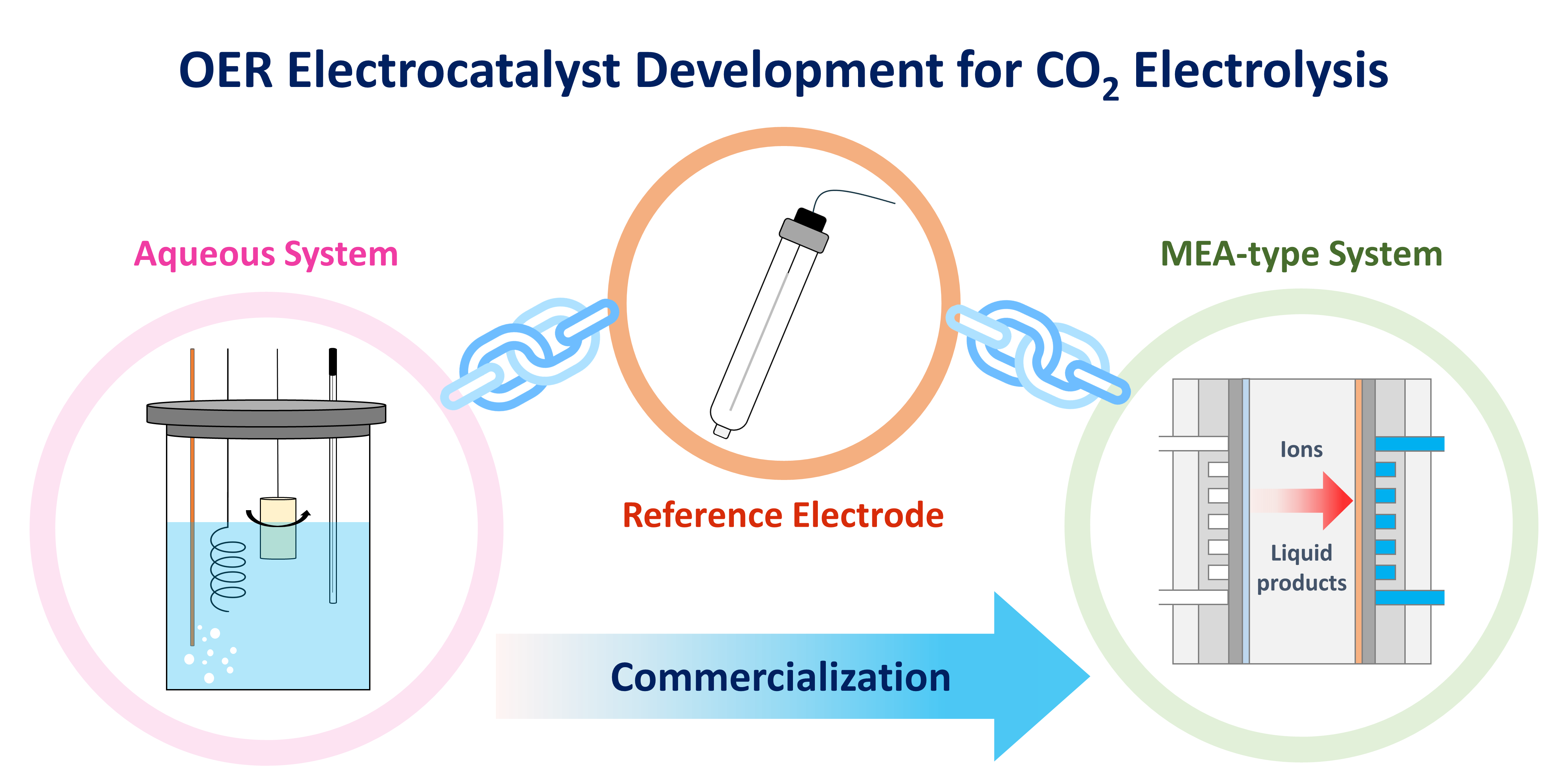
74. Seontaek Kwon, Tae-Hoon Kong, Namgyoo Park, Pandiarajan Thangavel, Hojeong Lee, Seokmin Shin, Jihoo Cha, Youngkook Kwon*
“Direction of Oxygen Evolution Reaction Electrocatalyst Evaluation for Anion Exchange Membrane CO2 Electrolyzer” , EES Catal., 2024, Link
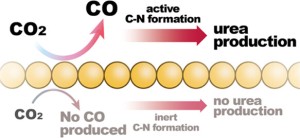
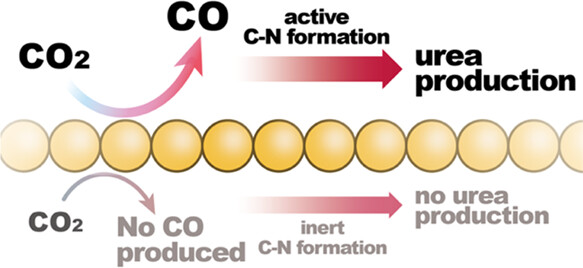
73. Hsien-Chin Li, Yeu-Shiuan Ho, Guo-Lin Yang, Ren-Han Li, Tung-Chun Kuo, Chi-Tien Hsieh, Youngkook Kwon, and Mu-Jeng Cheng*
“Linking CO to Urea Production from CO2 and NO3–/NO2– Co-Electrolysis on Transition Metals” , J. Phys. Chem. C, 2024, 128, 3, 1058-1067 Link
2023

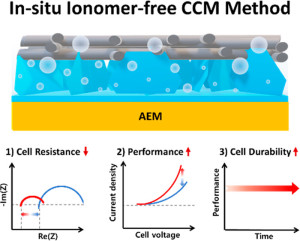 72. Tae-Hoon Kong, Pandiarajan Thangavel, Seokmin Shin, Seontaek Kwon, Hansaem Choi, Hojeong Lee, Namgyoo Park, Jung-Je Woo, Youngkook Kwon*
72. Tae-Hoon Kong, Pandiarajan Thangavel, Seokmin Shin, Seontaek Kwon, Hansaem Choi, Hojeong Lee, Namgyoo Park, Jung-Je Woo, Youngkook Kwon*
“In-Situ Ionomer-Free Catalyst-Coated Membranes for Anion Exchange Membrane Water Electrolyzers” , ACS Energy Lett., 2023, 8, 4666-4673 (Selected as Front Cover) Link and Cover Link

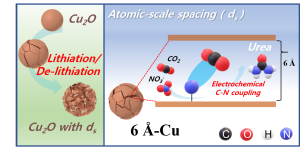
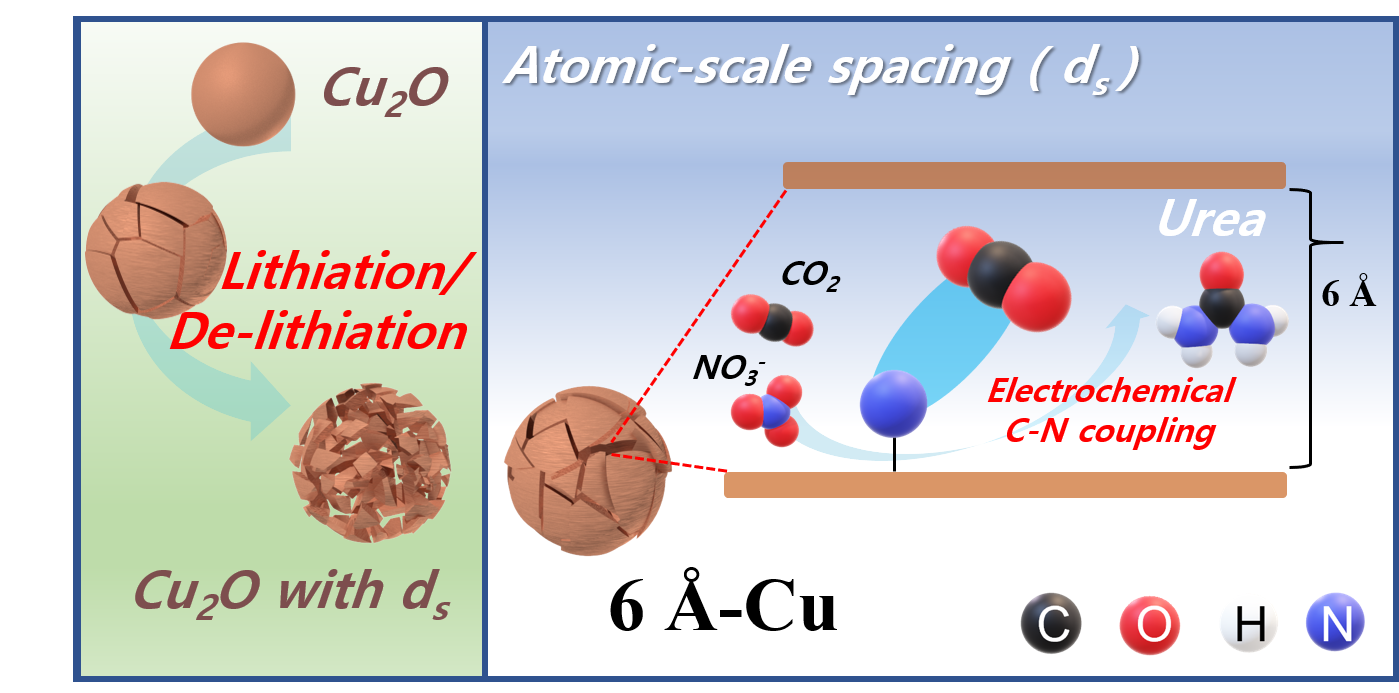
71. Seokmin Shin , Siraj Sultan , Zong-Xian Chen , Hojeong Lee , Hansaem Choi , Tae-Ung Wi , Changhyun Park , Tae Won Kim , Chanhee Lee , Jihong Jeong , Hyeju Shin , Tae-Hee Kim , Hyungkuk Ju , Hyung-Chul Yoon , Hyun-Kon Song* , Hyun-Wook Lee* , Mu-Jeng Cheng* and Youngkook Kwon*
“Copper with an atomic-scale spacing for efficient electrocatalytic co-reduction of carbon dioxide and nitrate to urea” , Energy Environ. Sci., 2023, 16, 2003-2013 (Selected as Inside back cover) Link and Cover Link

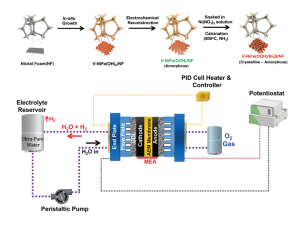 70. Pandiarajan Thangavel, Hojeong Lee, Tae-Hoon Kong, Seontaek Kwon, Ahmad Tayyebi, Ji-hoon Lee, Sung Mook Choi, Youngkook Kwon*
70. Pandiarajan Thangavel, Hojeong Lee, Tae-Hoon Kong, Seontaek Kwon, Ahmad Tayyebi, Ji-hoon Lee, Sung Mook Choi, Youngkook Kwon*
“Immobilizing Low-Cost Metal Nitrides in Electrochemically Reconstructed Platinum Group Metal (PGM)-Free Oxy-(Hydroxides) Surface for Exceptional OER Kinetics in Anion Exchange Membrane Water Electrolysis” , Adv. Energy Mater., 2023, 13, 2203401 (Selected as Front Cover) Link and Cover Link

69. Seokmin Shin, Tae-Ung Wi, Tae-Hoon Kong, Chanhyun Park, Hojeong Lee, Jihong Jeong, Eunryeol Lee, Subhin Yoon, Tae-Hee Kim, Hyun-Wook Lee, Youngkook Kwon*, Hyun-Kon Song*
“Selectively Enhanced Electrocatalytic Oxygen Evolution within Nanoscopic Channels Fitting a Specific Reaction Intermediate for Seawater Splitting“, Small., 2023, 19, 2206918 Link
68. Junghye Lee, Hansaem Choi, Jinhong Mun, Eunji Jin, Soochan Lee, Joohan Nam, Muhammad Umer, Jaeheung Cho, Geunsik Lee*, Youngkook Kwon*, Wonyoung Choe*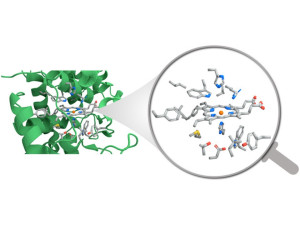

“Nanozyme Based on Porphyrinic Metal–Organic Framework for Electrocatalytic CO2 Reduction“, Small Structures, 2023, 4, 2200087 Link and Cover Link
2022
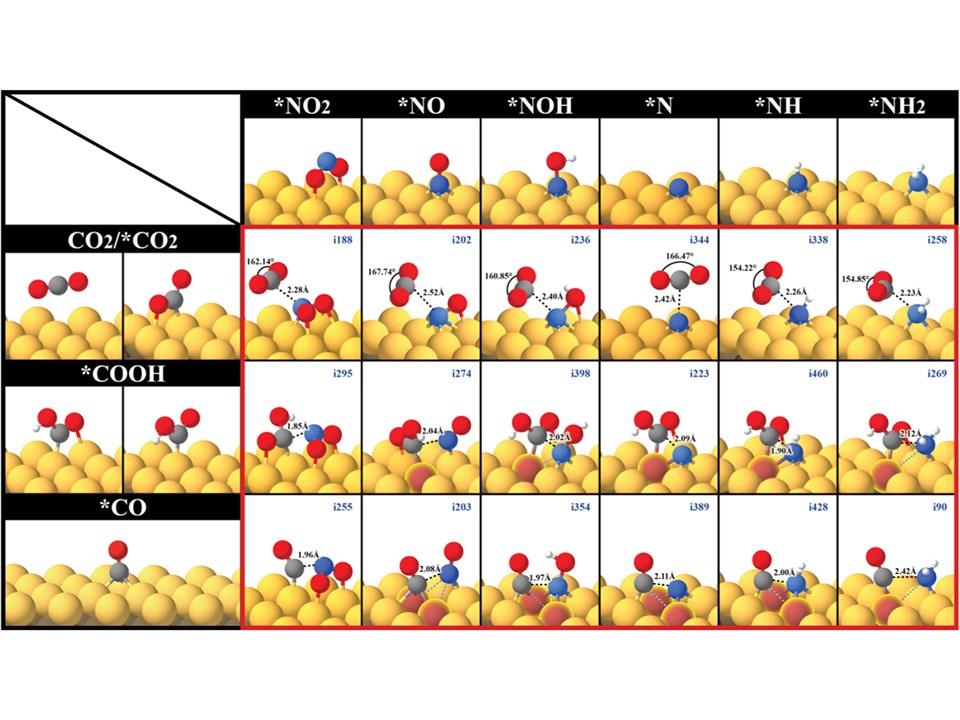
67. Guo-Lin Yang, Chi-Tien Hsieh, Yeu-Shiuan Ho, Tung-Chun Kuo, Youngkook Kwon, Qi Lu, and Mu-Jeng Cheng*
“Gaseous CO2 Coupling with N-Containing Intermediates for Key C-N Bond Formation during Urea Production from Coelectrolysis over Cu“, ACS Catal., 2022, 12, 18, 11494-11504 Link
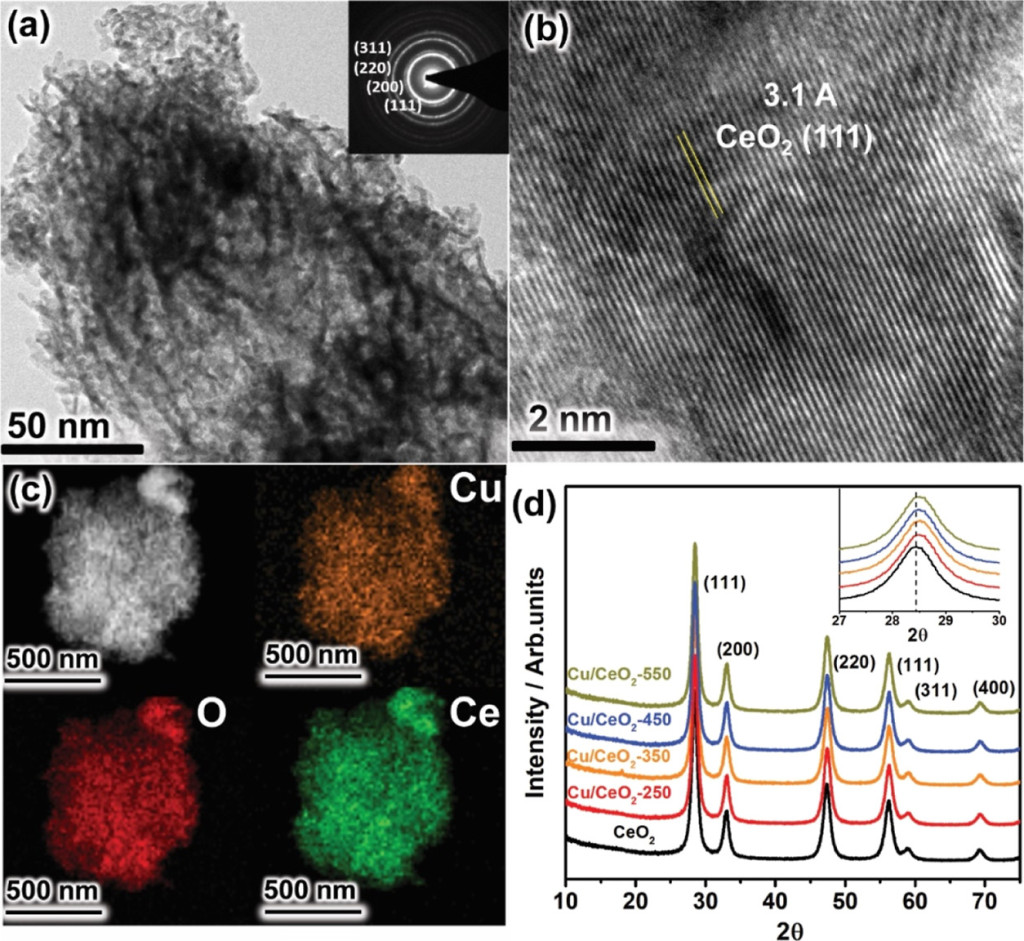
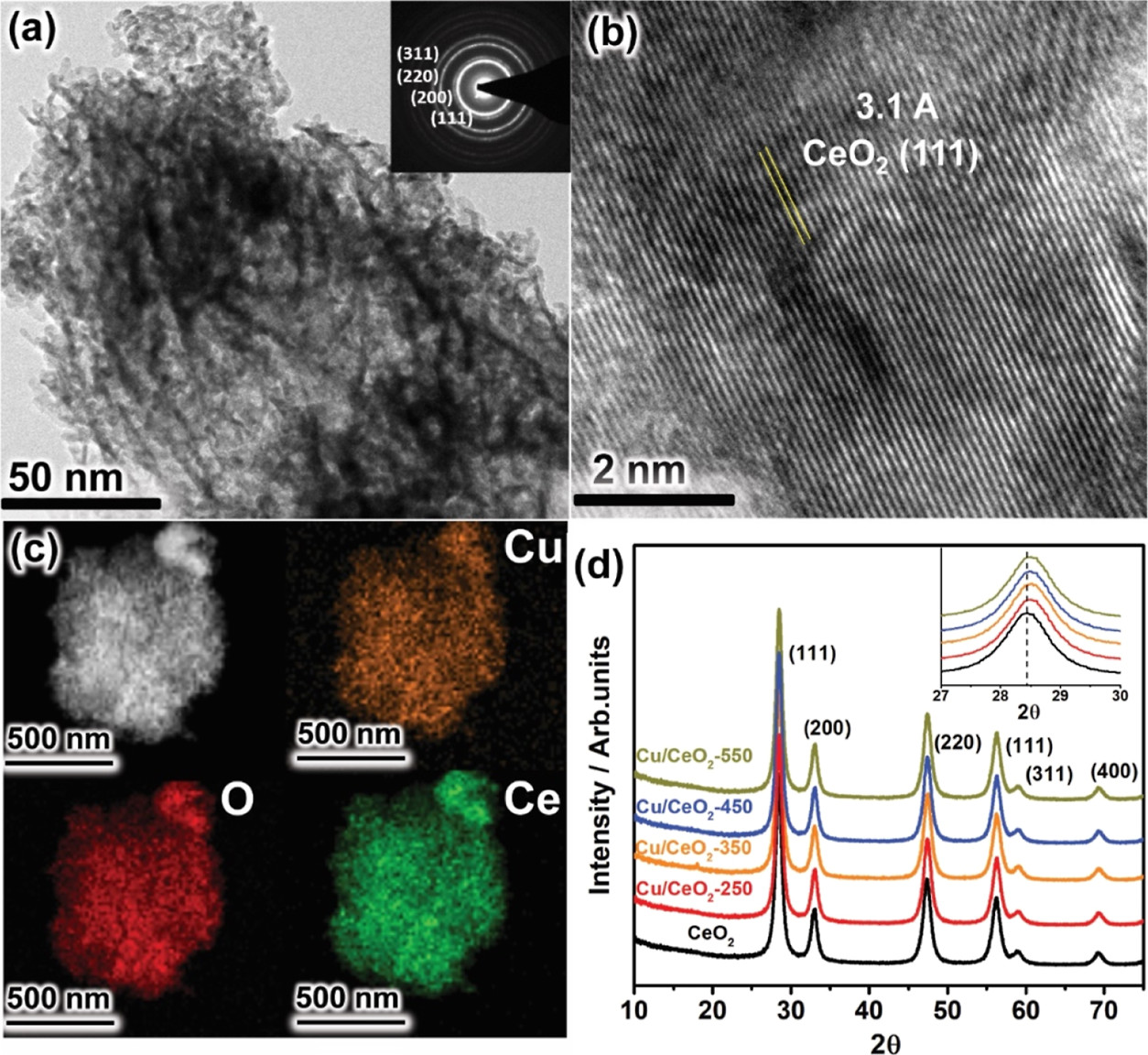
66. Kshirodra Kumar Patra, Zhu Liu, Hojeong Lee, Seungwon Hong, Hakhyeon Song, Hafiz Ghulam Abbas, Youngkook Kwon*, Stefan Ringe*, and Jihun Oh*
“Boosting Electrochemical CO2 Reduction to Methane via Tuning Oxygen Vacancy Concentration and Surface Termination on a Copper/Ceria Catalyst“, ACS Catal., 2022, 12, 17, 10973-10983 Link
65. Jae Bin Lee, Gun Ha Kim, Ji Hwan Jeon, Seo Yeong Jeong, Soochan Lee, Jaehyun Park, Doyoung Lee, Youngkook Kwon, Jeong Kon Seo, Joong-Hyun Chun, Seok Ju Kang, Wonyoung Choe, Jan-Uwe Rohde* & Sung You Hong*
“Rapid access to polycyclic N-heteroarenes from unactivated, simple azines via a base-promoted Minisci-type annulation“, Nat. Commun., 2022, 13(1) Link


64. Siraj Sultan‡, Hojeong Lee‡, Sojung Park‡, Minho M. Kim‡, Aram Yoon‡, Hansaem Choi, Tae-Hoon Kong, Young-Jin Koe, Hyung-Suk Oh, Zonghoon Lee*, Hyungjun Kim*, Wooyul Kim* and Youngkook Kwon*
“Interface rich CuO/Al₂CuO₄ surface for selective ethylene production from electrochemical CO₂ conversion“, Energy Environ. Sci., 2022, 15, 2397-2409 (Selected as Inside Front Cover) Link and Cover Link
63. Seonjeong Cheon, Won June Kim, Dong Yeon Kim*, Youngkook Kwon*, and Jong-In Han*
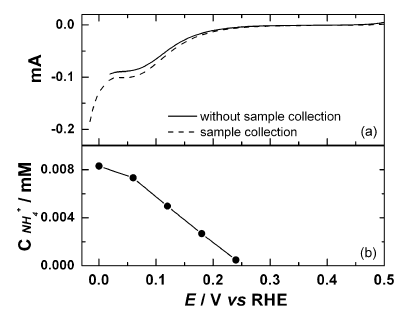
“Electro-synthesis of Ammonia from Dilute Nitric Oxide on a Gas Diffusion Electrode“, ACS Energy Lett., 2022, 7, 3, 958-965 Link
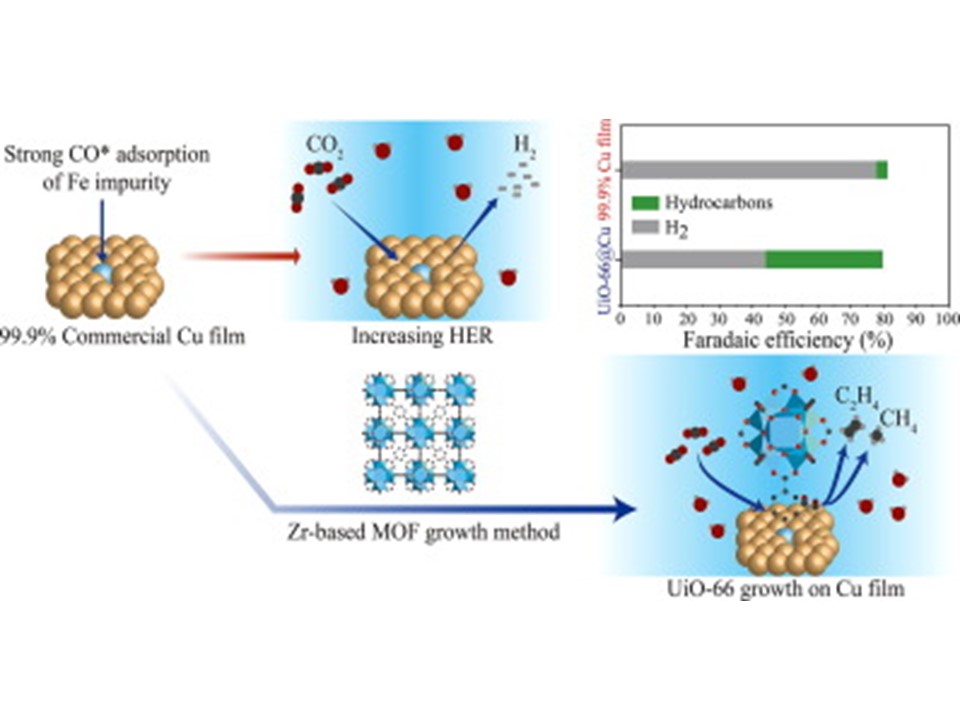
62. Shin Joon Kang, Jong Ho Won, Hansaem Choi, Woohyeong Sim, Mun Kyoung Kim, Siraj Sultan, Youngkook Kwon*, Hyung Mo Jeong*
“Compensating the impurities on the Cu surface by MOFs for enhanced hydrocarbon production in the electrochemical reduction of carbon dioxide“, J. Energy Chem., 2022, 66, 68-73 Link
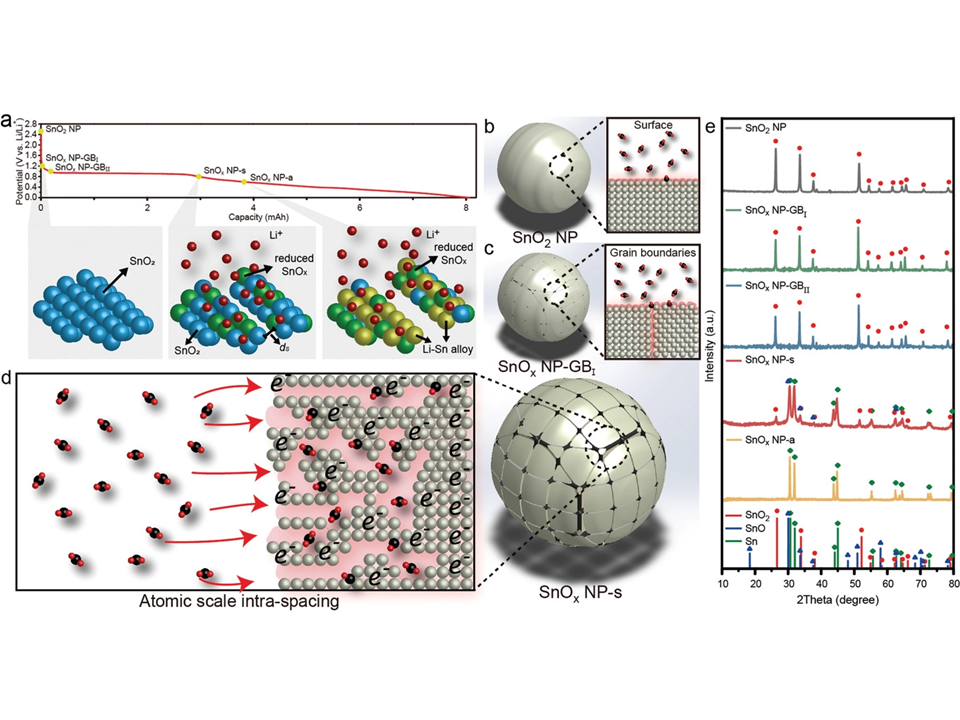

61. Mun Kyoung Kim, Hojeong Lee, Jong Ho Won, Woohyeong Sim, Shin Joon Kang, Hansaem Choi, Monika Sharma, Hyung-Suk Oh, Stefan Ringe,* Youngkook Kwon,* and Hyung Mo Jeong**
“Design of less than 1 nm Scale Spaces on SnO₂ Nanoparticles for High-Performance Electrochemical CO₂ Reduction“, Adv. Funct. Mater., 2022, 32, 8, (Selected as Front Cover) Link and Cover Link
2021
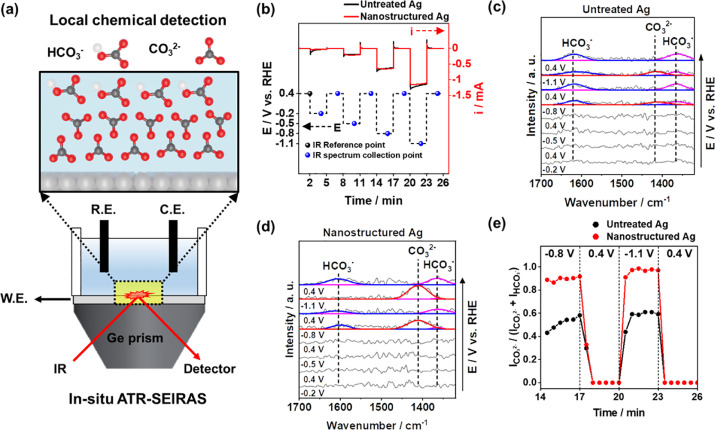
60. Jungae Lim, Hyungseob Lim, Bupmo Kim, Soo Min Kim, Jong-Bae Lee, Kang Rae Cho, Hansaem Choi, Siraj Sultan, Wonyong Choi, Wooyul Kim*, Youngkook Kwon*
“Local pH induced electrochemical CO₂ reduction on nanostructured Ag for adjustable syngas composition“, Electrochim. Acta, 2021, 395, 1, 139190 Link
59. Siraj Sultan, Jin Hyun Kim, SeungHyeon Kim, Youngkook Kwon*, Jae Sung Lee*
“Innovative strategies toward challenges in PV-powered electrochemical CO2 reduction“, J. Energy. Chem., 2021, 60, 410-416 Link
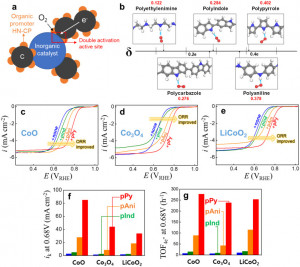
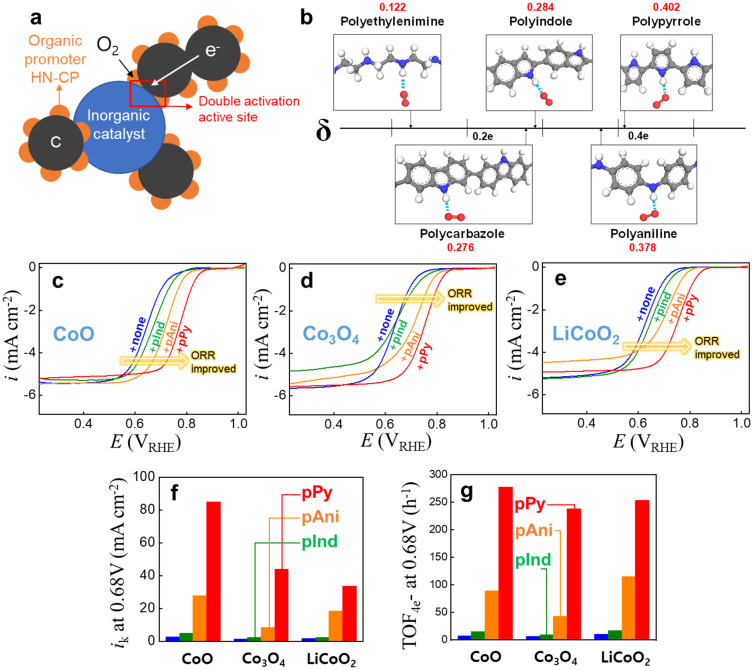
58. Dong-Gyu Lee, Su Hwan Kim, Jiyun Lee, Seokmin Shin, Se Hun Joo, Yeongdae Lee, Chanhyun Park, Youngkook Kwon, Sang Kyu Kwak*, Hyun-Kon Song*
“Double activation of oxygen intermediates of oxygen reduction reaction by dual inorganic/organic hybrid electrocatalysts“, Nano Energy., 2021, 86, 106048 Link
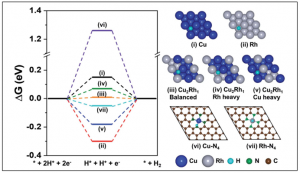
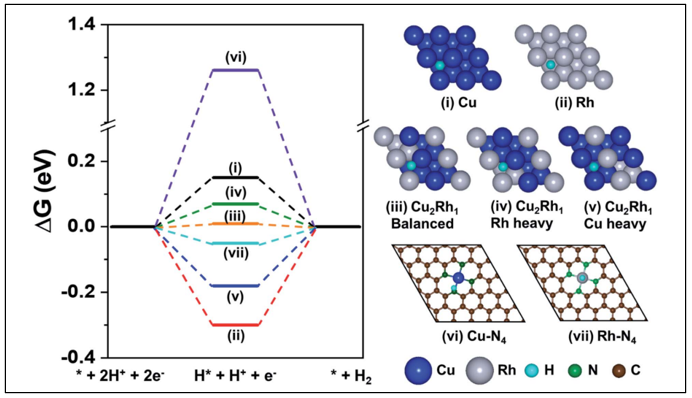
57. Siraj Sultan, Muhammad Hanif Diorizky, Miran Ha, Jitendra N. Tiwari, Hansaem Choi, Ngoc Kim Dang, Pandiarajan Thangavel, Jong Hoon Lee, Hu Young Jeong, Hyeon Suk Shin, * Youngkook Kwon * and Kwang S. Kim*
“Modulation of Cu and Rh single-atoms and nanoparticles for high-performance hydrogen evolution activity in acidic media“, JMCA ., 2021, 9, 10326 Link
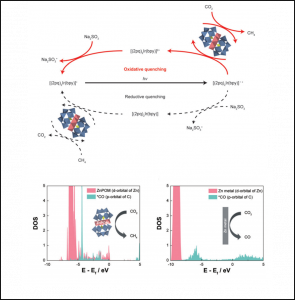
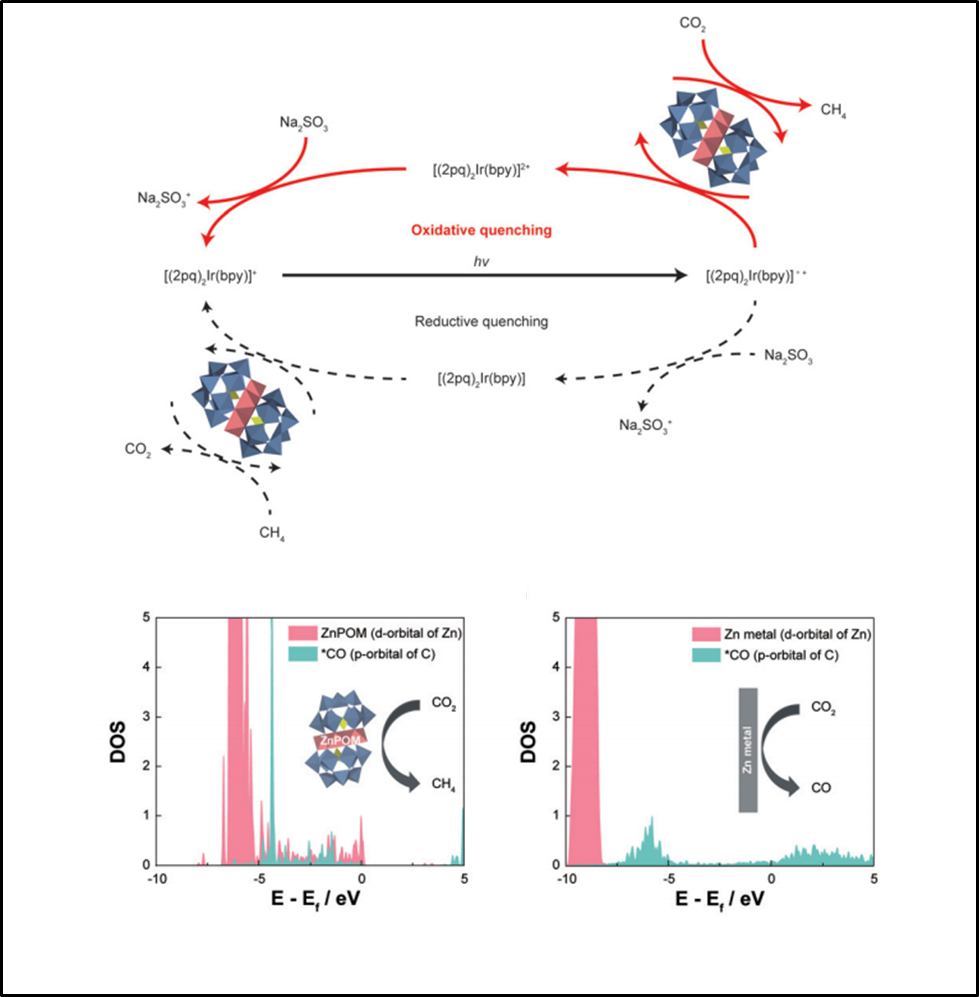
56. Nayeong Kim, Jung Seung Nam, Jinhyung Jo, Junmo Seong, Hyunwoo Kim, Youngkook Kwon, Myoung Soo Lah, Jun Hee Lee*, Tae-Hyuk Kwon*, Jungki Ryu*
“Selective Photocatalytic Production of CH4 Using Zn-based Polyoxometalate as a Nonconventional CO2 Reduction Catalyst“, Nanoscale Horizons., 2021, 6, 379-385 Link

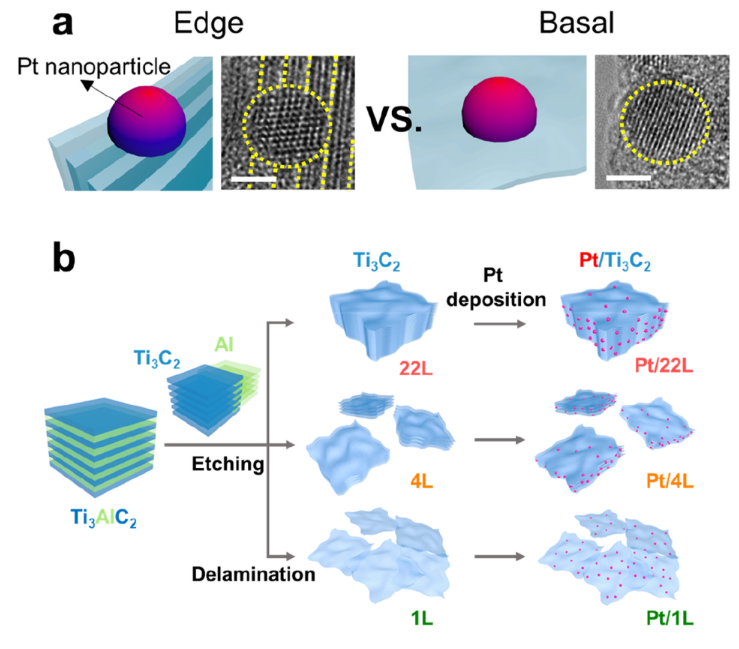
55. Yeongdae Lee, Jang Hyuk Ahn, Hee-Young Park, Jeawoo Jung, Yuju Jeon, Dong-Gyu Lee, Min-Ho Kim, Eunryeol Lee, Chanseok Kim, Youngkook Kwon, Hyun-Wook Lee, Jong Hyun Jang, Jun Hee Lee, Hyun-Kon Song*
“Support structure-catalyst electroactivity relation for oxygen reduction reaction on platinum supported by two-dimensional titanium carbide“, Nano Energy., 2021, 79, 105363 Link
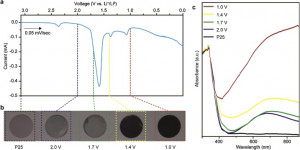
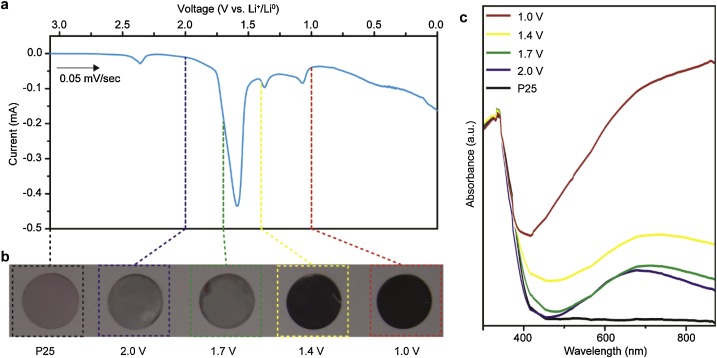
54. Mun Kyoung Kim, Woo Hyeong Sim, Miri Choi, Hyungseob Lim, Youngkook Kwon*, Hyung Mo Jeong*
“Electrochemically Li-intercalated TiO2 nanoparticles for High performance photocatalytic production of hydrogen“, Catal. Today., 2021, 1, 23-27 Link
2020
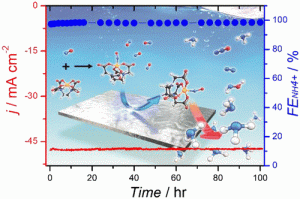
53. DongYeon Kim, Dongyup Shin, Juheon Heo, Hyungseob Lim, Jung-Ae Lim, Hyung Mo Jeong, Beom-Sik Kim, Iljeong Heo, Inhwan Oh, Boreum Lee, Monika Sharma, Hankwon Lim*, Hyungjun Kim*, and Youngkook Kwon*
“Unveiling Electrode–Electrolyte Design-Based NO Reduction for NH3 Synthesis“, ACS Energy Lett., 2020, 11, 3647-3656 Link
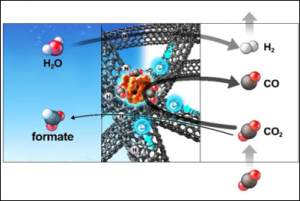
52. Mi-Young Lee, Stefan Ringe, Hyungjun Kim*, Seoktae Kang*, and Youngkook Kwon*
“Electric field mediated selectivity switching of electrochemical CO2 reduction from formate to CO on carbon supported Sn“, ACS Energy Lett., 2020, 9, 2987-2994 Link
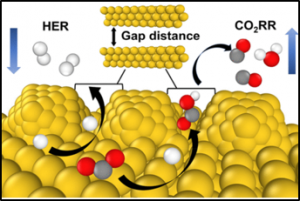
51. Kuan Chang, Xianfeng Jian, HyungMo Jeong, Youngkook Kwon, Qi Lu* and Mu-Jeng Cheng*
“Improving CO2 Electrochemical Reduction to CO Using Space Confinement between Gold or Silver Nanoparticles“, J. Phys. Chem. Lett., 2020, 11, 1896-1902 Link

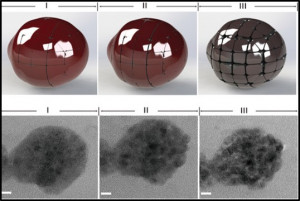
50. Youngkook Kwon+, Hyung Mo Jeong+, Jong Ho Won, Yanwei Lum, Mu-Jeng Cheng, Kwang Ho Kim, Martin Head-Gordon, and Jeung Ku Kang* (+: equal contribution)
“Atomic-Scale Spacing between Copper Facets for the Electrochemical Reduction of Carbon Dioxide“, Adv. Energy Mater., 2020, 10, 1903423. (Selected as Front Cover) Link and Cover Link
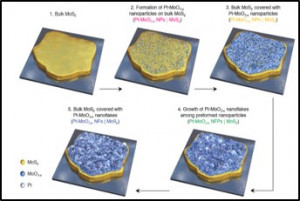
49. Daewon Lee, Youngmin Kim, Hyun Woo Kim, Min Choi, Noejung Park, Hyunju Chang, Youngkook Kwon, Jong Hyeok Park, Hyung Ju Kim*
“In situ electrochemically synthesized Pt-MoO3−x nanostructure catalysts for efficient hydrogen evolution reaction“, J. Catal., 2020, 381, 1-13. Link

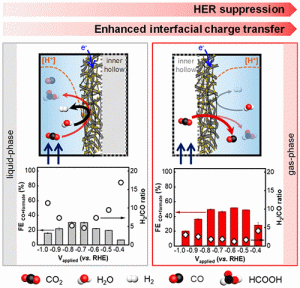
48. Mi-Young Lee, Seungyeob Han, Hyungseob Lim, Youngkook Kwon*, Seoktae Kang*
“Electrocatalytic CO2 Reduction via a Permeable CNT Hollow-Fiber Electrode Incorporated with SnO2 Nanoparticles“, ACS Sustainable Chem. Eng., 2020, 8, 5, 2117–2121. (Selected as Cover) Link and Cover Link
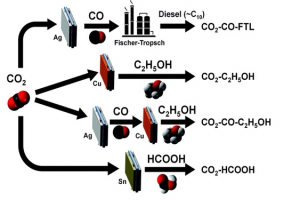
47. Mi-Young Lee, Ki Tae Park, Wonhee Lee, Hyungseob Lim, Youngkook Kwon*, Seoktae Kang*
“Current achievements and the future direction of electrochemical CO2 reduction: A short review“, Crit. Rev. Environ. Sci. Technol., 2020, 50.8: 769-815. Link
2019
46. Mun Kyoung Kim, Woo Hyeong Sim, Miri Choi, Hyungseob Lim, Youngkook Kwon*, Hyung Mo Jeong*
“Electrochemically Li-intercalated TiO2 nanoparticles for High performance photocatalytic production of hydrogen“, Catal. Today., 2021, 1, 23-27 Link (2019 Available online)
2019 - Before UNIST

45. Mun Kyoung Kim, Hyeok Joo Kim, Hyungseob Lim, Youngkook Kwon*, Hyung Mo Jeong*
“Metal–organic framework-mediated strategy for enhanced methane production on copper nanoparticles in electrochemical CO2 reduction“, Electrochim. Acta., 2019, 306, 28-34. Link
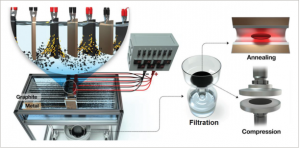
44. Yeon Ju Kwon, Youngkook Kwon, Ho Seok Park,* and Jea Uk Lee*
“Mass-Produced Electrochemically Exfoliated Graphene for Ultrahigh Thermally Conductive Paper Using a Multimetal Electrode System“, Adv. Mater. Interfaces., 2019, 6, 1900095 Link
2018
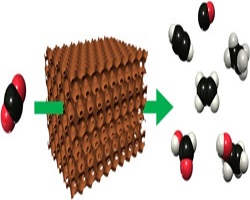
43. Hakhyeon Song, Mintaek Im, Jun Tae Song, Jung-Ae Lim, Beom-Sik Kim, Youngkook Kwon*, Sangwoo Ryu*, Jihun Oh*
“Effect of mass transfer and kinetics in ordered Cu-mesostructures for electrochemical CO2 reduction“, Appl. Catal. B: Environ., 2018, 232, 391-396 Link

42. Hyung Mo Jeong, Boon Siang Yeo, Youngkook Kwon*
“Copper Catalysts for the Electrochemical Reduction of Carbon Dioxide“, Electrochemical Reduction of Carbon Dioxide: Overcoming the Limitations of Photosynthesis. Ed. by David J. Fermin and Frank Marken, 2018, p.63-87. Link
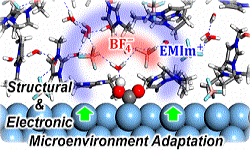
41. Hyung-Kyu Lim, Youngkook Kwon, Han Seul Kim, Jiwon Jeon, Yong-Hoon Kim, Jung-Ae Lim, Beom-Sik Kim, Jina Choi*, Hyungjun Kim*
“Insight into the Microenvironments of the Metal-Ionic Liquid Interface during Electorchemical CO2 Reduction“, ACS Catal., 2018, 8, 3. Link
2017
40. Hyung-Kyu Lim, Youngkook Kwon, Han Seul Kim, Jiwon Jeon, Yong-Hoon Kim, Jung-Ae Lim, Beom-Sik Kim, Jina Choi*, Hyungjun Kim*
“Quadruple metal-based layered structure as the photocatalyst for conversion of carbon dioxide into a value added carbon monoxide with high selectivity and efficiency“, J. Mater. Chem. A, 2017, 5. Link

39. Youngkook Kwon
“전기화학기반 화학원료 생산“, 녹색기후기술백서, 2017
2016
38. Meenesh R. Singh, Youngkook Kwon, Yanwei Lum, Joel W. Ager, and Alexis T. Bell*
“Hydrolysis of Electrolyte Cations Enhances the Electrochemical Reduction of CO2 over Ag and Cu“, J. Am. Chem. Soc., 2016, 138, 39. Link
37. Youngkook Kwon, Klaas Jan P Schouten, Jan C van der Waal, Ed de Jong, Marc TM Koper*
“Electrocatalytic conversion of furanic compounds“, ACS Catal., 2016, 6, 10, 6704-6717. Link
36. Amanda C Garcia, Manuel J Kolb, Chris van Nierop y Sanchez, Jan Vos, Yuvraj Y Birdja, Youngkook Kwon, Germano Tremiliosi-Filho, Marc TM Koper*
“Strong impact of platinum surface structure on primary and secondary alcohol oxidation during electro-oxidation of glycerol“, ACS Catal., 2016, 6, 7. Link
35. Youngkook Kwon, Yanwei Lum, Ezra L Clark, Joel W Ager*, Alexis T Bell*
“CO2 Electroreduction with Enhanced Ethylene and Ethanol Selectivity by Nanostructuring Polycrystalline Copper“, ChemElectroChem, 2016, 3, 1012–1019. Link
34. Peter Lobaccaro, Meenesh R Singh, Ezra Lee Clark, Youngkook Kwon, Alexis T Bell*, Joel W Ager*
“Effects of temperature and gas–liquid mass transfer on the operation of small electrochemical cells for the quantitative evaluation of CO2 reduction electrocatalysts“, Phys. Chem. Chem. Phys., 2016, 18, 26777-26785. Link
33. R. Kortlever,* I. Peters, C. Balemans, R. Kas, Y. Kwon, G. Mulb and M. T. M. Koper*
“Palladium–gold catalyst for the electrochemical reduction of CO2 to C1–C5 hydrocarbons“, Chem. Commun., 2016, 52, 10229-10232. Link
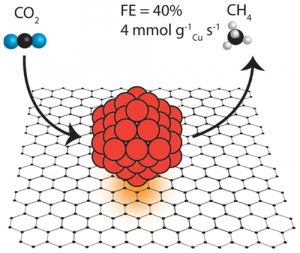
32. Yanwei Lum+, Youngkook Kwon+, Peter Lobaccaro, Le Chen, Ezra Lee Clark, Alexis T Bell, Joel W Ager (+: equal contribution)
“Trace Levels of Copper in Carbon Materials Show Significant Electrochemical CO2 Reduction Activity“, ACS Catal., 2016, 6, 202-209. Link
2015
31. Mu-Jeng Cheng, Youngkook Kwon, Martin Head-Gordon, Alexis T Bell
“Tailoring Metal-Porphyrin-Like Active Sites on Graphene to Improve the Efficiency and Selectivity of Electrochemical CO2 Reduction“, J. Phys. Chem. C, 2015, 119, 37. Link
30. Jing Shen, Ruud Kortlever, Recep Kas, Yuvraj Y Birdja, Oscar Diaz-Morales, Youngkook Kwon, Isis Ledezma-Yanez, Klaas Jan P Schouten, Guido Mul, Marc TM Koper
“Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoporphyrin“, Nat. Commun., 2015, 6, 8177. Link
29. Ezra L Clark, Meenesh R Singh, Youngkook Kwon, Alexis T Bell
“Differential Electrochemical Mass Spectrometer Cell Design for Online Quantification of Products Produced during Electrochemical Reduction of CO2“, Anal. Chem., 2015, 87, 15, 8013-8020. Link
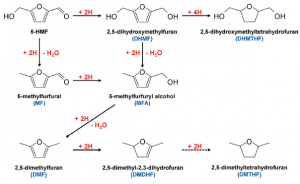
28. Youngkook Kwon, Yuvraj Y Birdja, Saeed Raoufmoghaddam, Marc TM Koper
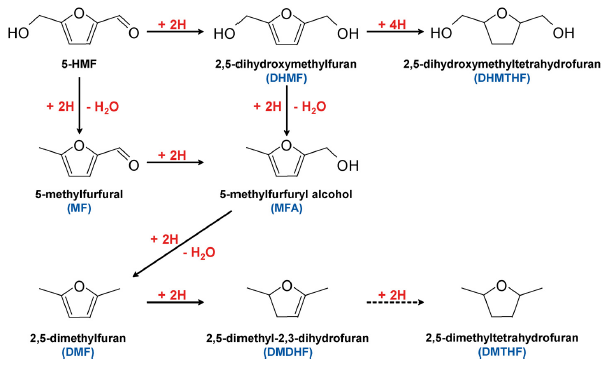
“Electrocatalytic Hydrogenation of 5‐Hydroxymethylfurfural in Acidic Solution“, ChemSusChem, 2015, 8, 1745-1751. Link
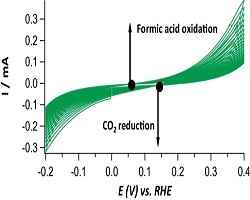
27. Ruud Kortlever, Collin Balemans, Youngkook Kwon, Marc TM Koper
“Electrochemical CO2 reduction to formic acid on a Pd-based formic acid oxidation catalyst“, Catal. Tod., 2015, 244, 58-62. Link
26. Youngkook Kwon, Ed de Jong, Jan Kees van der Waal, Marc TM Koper
“Selective electrocatalytic oxidation of sorbitol to fructose and sorbose“, ChemSusChem, 2015, 8, 970-973. Link
2014
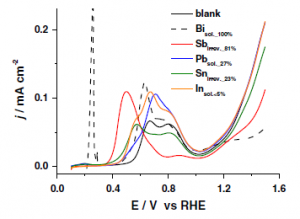
25. Youngkook Kwon, Thomas JP Hersbach, Marc TM Koper
“Electro-oxidation of glycerol on platinum modified by adatoms: activity and selectivity effects“, Top. Catal., 2014, 57, 1272-1276. Link
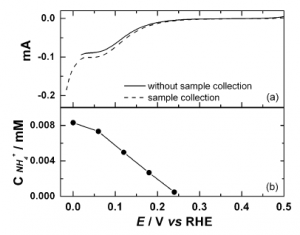
24. W Siriwatcharapiboon, Y Kwon, J Yang, RL Chantry, Z Li, SL Horswell, MTM Koper
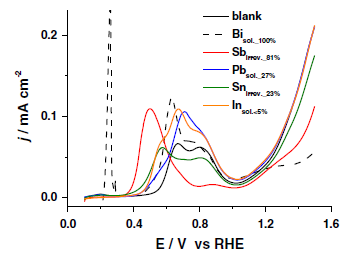
“Promotion effects of Sn on the electrocatalytic reduction of nitrate at Rh nanoparticles“, ChemElectroChem., 2014, 1, 172-179. Link
23. Youngkook Kwon, Stefan J Raaijman, Marc TM Koper
“Role of peroxide in the catalytic activity of gold for oxidation reactions in aqueous media: an electrochemical study“, ChemCatChem, 2014, 6, 79-81. Link
2013
22. Hongjiao Li, Federico Calle-Vallejo, Manuel J Kolb, Youngkook Kwon, Yongdan Li, Marc TM Koper
“Why (1 0 0) Terraces Break and Make Bonds: Oxidation of Dimethyl Ether on Platinum Single-Crystal Electrodes“, J. Am. Chem. Soc., 2013, 135, 38, 14329-14338. Link
21. Youngkook Kwon, Ed de Jong, Saeed Raoufmoghaddam, Marc TM Koper
“Electrocatalytic Hydrogenation of 5‐Hydroxymethylfurfural in the Absence and Presence of Glucose“, ChemSusChem, 2013, 6, 1659-1667 . Link
20. Jian Yang, Youngkook Kwon, Matteo Duca, Marc TM Koper
“Combining voltammetry and ion chromatography: application to the selective reduction of nitrate on Pt and PtSn electrodes“, Anal. Chem., 2013, 85, 16, 7645-7649. Link
19. R Kortlever, KH Tan, Y Kwon, MTM Koper
“Electrochemical carbon dioxide and bicarbonate reduction on copper in weakly alkaline media“, J. Solid State Electrochem., 2013, 17, 1843–1849. Link
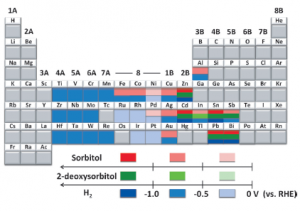
18. Youngkook Kwon, Marc TM Koper
“Electrocatalytic hydrogenation and deoxygenation of glucose on solid metal electrodes“, ChemSusChem, 2013, 6, 455-462. Link
2012
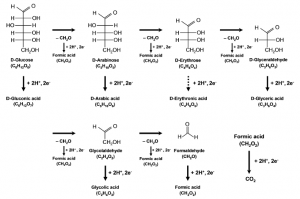
17. Youngkook Kwon, Steven EF Kleijn, Klaas Jan P Schouten, Marc TM Koper
“Cellobiose hydrolysis and decomposition by electrochemical generation of acid and hydroxyl radicals“, ChemSusChem, 2012, 5, 1935-1943. Link
16. Youngkook Kwon, Yuvraj Birdja, Ioannis Spanos, Paramaconi Rodriguez, Marc TM Koper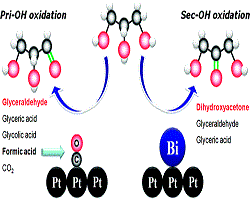
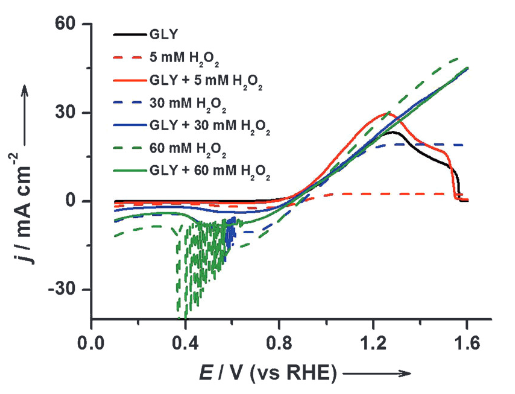
“Highly selective electro-oxidation of glycerol to dihydroxyacetone on platinum in the presence of bismuth“, ACS Catal., 2012, 2.5, 759-764. Link
15. Paramaconi Rodriguez, Youngkook Kwon, Marc TM Koper

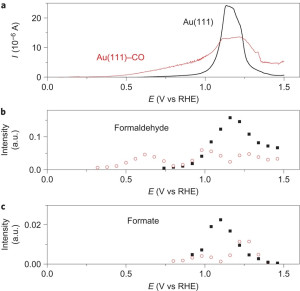
“The promoting effect of adsorbed carbon monoxide on the oxidation of alcohols on a gold catalyst“, Nature Chemistry, 2012, 4, 172-182. (Selected as Cover) Link and Cover Link
2011
14. Youngkook Kwon, Klass Jan P Schouten, Marc TM Koper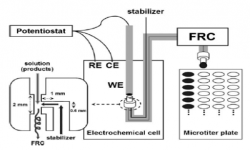
“Mechanism of the catalytic oxidation of glycerol on polycrystalline gold and platinum electrodes“, ChemCatChem, 2011, 3, 1176-1185. Link
13. Annukka Santasalo-Aarnio, Youngkook Kwon, Elisabet Ahlberg, Kyosti Kontturi, Tanja Kallio, Marc TM Koper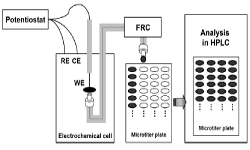
“Comparison of methanol, ethanol and iso-propanol oxidation on Pt and Pd electrodes in alkaline media studied by HPLC“, Electrochem. Commun., 2011, 13, 466-469. Link
12. Youngkook Kwon, Stanley CS Lai, Paramaconi Rodriguez, Marc TM Koper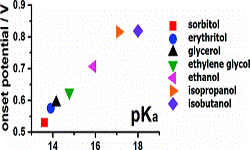
“Electrocatalytic oxidation of alcohols on gold in alkaline media: base or gold catalysis?“, J. Am. Chem. Soc., 2011, 133, 6914-6917. Link
11. Youngkook Kwon, Hyejin Lee, Jaeyoung Lee

“Autonomous interfacial creation of nanostructured lead oxide“, Nanoscale, 2011, 3, 4984-4988. (Selected as Front Cover) Link and Cover Link
10. KJP Schouten, Youngkook Kwon, CJM Van der Ham, Z Qin, Marc TM Koper
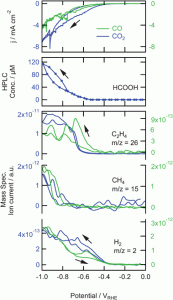
“A new mechanism for the selectivity to C1 and C2 species in the electrochemical reduction of carbon dioxide on copper electrodes“, Chem. Sci., 2011, 2, 1902-1909. (Selected as Inside Front Cover) Link and Cover Link
2010
9. Jy Joo, JK Lee, Y Kwon, CR Jung, ES Lee, JH Jang, HJ Lee, S Uhm, J Lee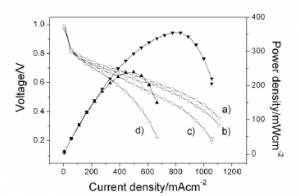
“Enhancement of CO Tolerance on Electrodeposited Pt Anode for Micro-PEM Fuel Cells“, Fuel Cells, 2010, 10, 926-931. Link
8. Hongrae Jeon, Jiyong Joo, Youngkook Kwon, Sunghyun Uhm, Jaeyoung Lee

“Morphological features of electrodeposited Pt nanoparticles and its application as anode catalysts in polymer electrolyte formic acid fuel cells“, J. Power Sources, 2010, 195, 5929-5933. Link
7. Youngkook Kwon, Jaeyoung Lee
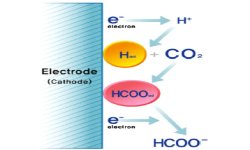
“Formic acid from carbon dioxide on nanolayered electrocatalyst“, Electrocatalysis, 2010, 1, 108-115. Link
6. Youngkook Kwon, Marc TM Koper
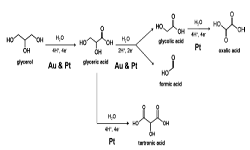
“Combining voltammetry with HPLC: application to electro-oxidation of glycerol“, Anal. Chem, 2010, 82, 13, 5420-5424. Link
2009
5. Jaeyoung Lee, Youngkook Kwon, Revocatus L Machunda, Hye Jin Lee
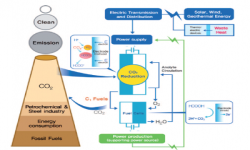
“Electrocatalytic recycling of CO2 and small organic molecules“, Chemistry – An Asian Journal, 2009, 4, 1516-1523. Link
4. Youngkook Kwon, Jae-Kwang Lee, Duk-Jin Ji, Jae-Young Lee
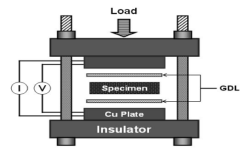
“Electrochemical Characteristics of Home-Made Bipolar Plate and Its Relationship with Fuel Cell Performance“, JKES, 2009, 12, 1, 68-74. Link
2008
3. Sunghyun Uhm, Hye Jin Lee, Youngkook Kwon, Jaeyoung Lee
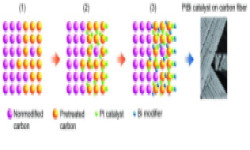
“A Stable and Cost-Effective Anode Catalyst Structure for Formic Acid Fuel Cells“, Angew. Chem. Int. Ed., 2008, 47, 52, 10163-10166. Link
2. Sunghyun Uhm, Hye Jin Lee, Youngkook Kwon, Jaeyoung Lee
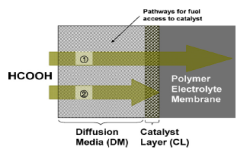
“Highly effective anode structure in a direct formic acid fuel cell“, Electrochim. Acta, 2008, 53, 16, 5162-5168. Link
1. Youngkook Kwon, Sung-Hyun Uhm, Jae-Young Lee
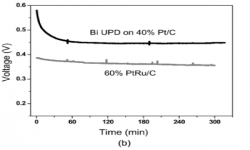
“Development of a Formic Acid Fuel Cell Anode by Multi-layered Bismuth Modification“, Korean Chemical Engineering Research, 2008, 46, 4, 697-700. Link








{kind=link}